

|
Protein Structure Prediction
|
The Critical Assessment of Structure Prediction (CASP) is a community-wide experiment, which designs to benchmark the state-of-the-art of protein structure prediction in every two years since 1994. Our lab has participated as "Zhang-Server" in the automated structure prediction section since 2006, which the method has been consistently ranked at the top in the experiments (Table 1). The results of recent CASP experiments can be found at Protein Structure Prediction Center.

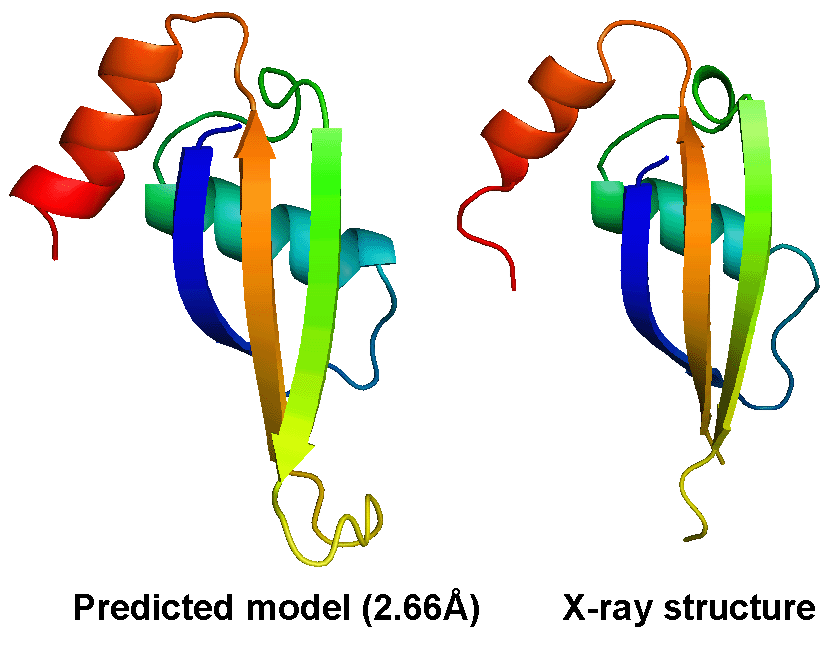
The most difficult problem in protein structure prediction is the modeling of proteins which have no solved structures that can be used as template, commonly referred "ab initio" or "free modeling (FM)" modeling. Figure 1 shows a successful example of ab initio modeling on a FM target (T0604_1) in CASP9, where the first model by the I-TASSER server has a RMSD 2.66 Angstroms to the X-ray crystal structure.
Despite the successes, significant unsolved problems remain in protein structure prediction, which will be the target of our lab in the next few years. These include:
|
Protein Design
|
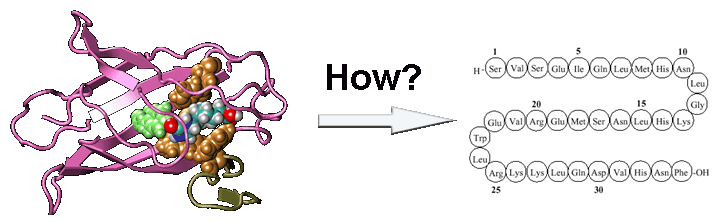
We successfully designed a number of new protein sequences based on a physics-based atomic force field with the lowest free-energy state searched by Monte Carlo simulation, followed by sequence-based clustering. The designed protein sequence can be folded by I-TASSER with a RMSD <2 Angstroms in 62% of cases, despite that the I-TASSER force field differs significantly from that used in the design. Figure 3 shows three representative examples of the target protein structure and I-TASSER model of the designed sequences.

Figure 3.
I-TASSER models of design sequences (red) versus crystal structure of target proteins (green)
for calcium-binding domain of Calx (3E9TA), odorant binding protein (2ERBA), and peptidyl-tRNA
hydrolase (1WN2A). The sequence identities of the designed and target sequences are all below 30%.
Recently, we proposed a new protocol, EvoDesign, which uses evolutionary profiles to guide folding refinement of new designs, with biological functions introduced by protein-interface binding profiles and interactions. The protocol was recently used to successfully design functional XIAP (X-linked Inhibitor of Apoptosis Protein) BIR3 domains capable of binding Smac peptides but not inhibiting caspase-9 proteolytic activity in vitro, which demonstrated the potential to change apoptosis pathways through computational protein design (Figure 4).
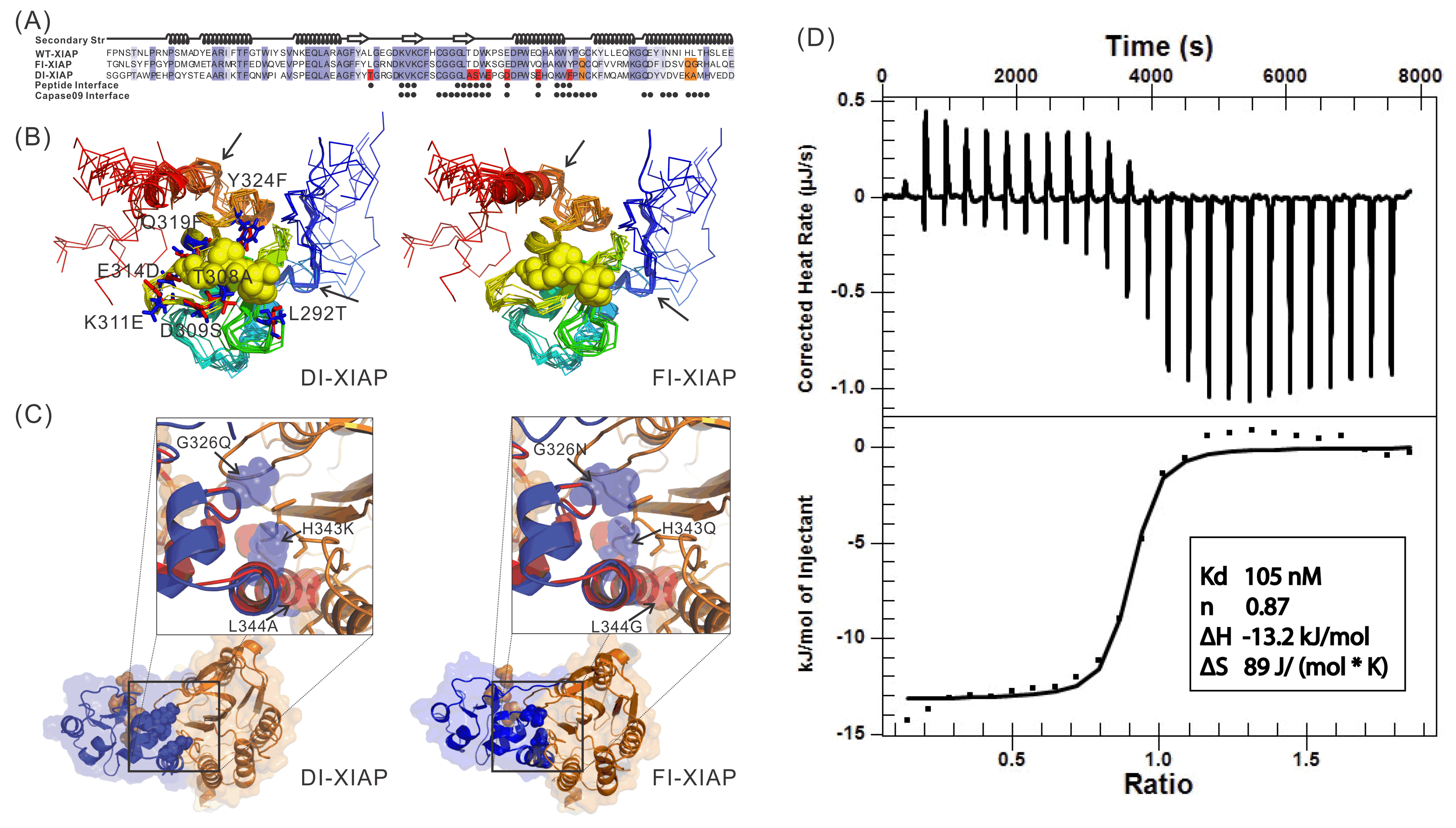
Figure 4.
Sequence and structure of two XIAPs designed by EvoDesign which binds with
Smac peptides but not inhibiting caspase-9 proteolytic activity in vitro.
|
Protein Function Prediction
|

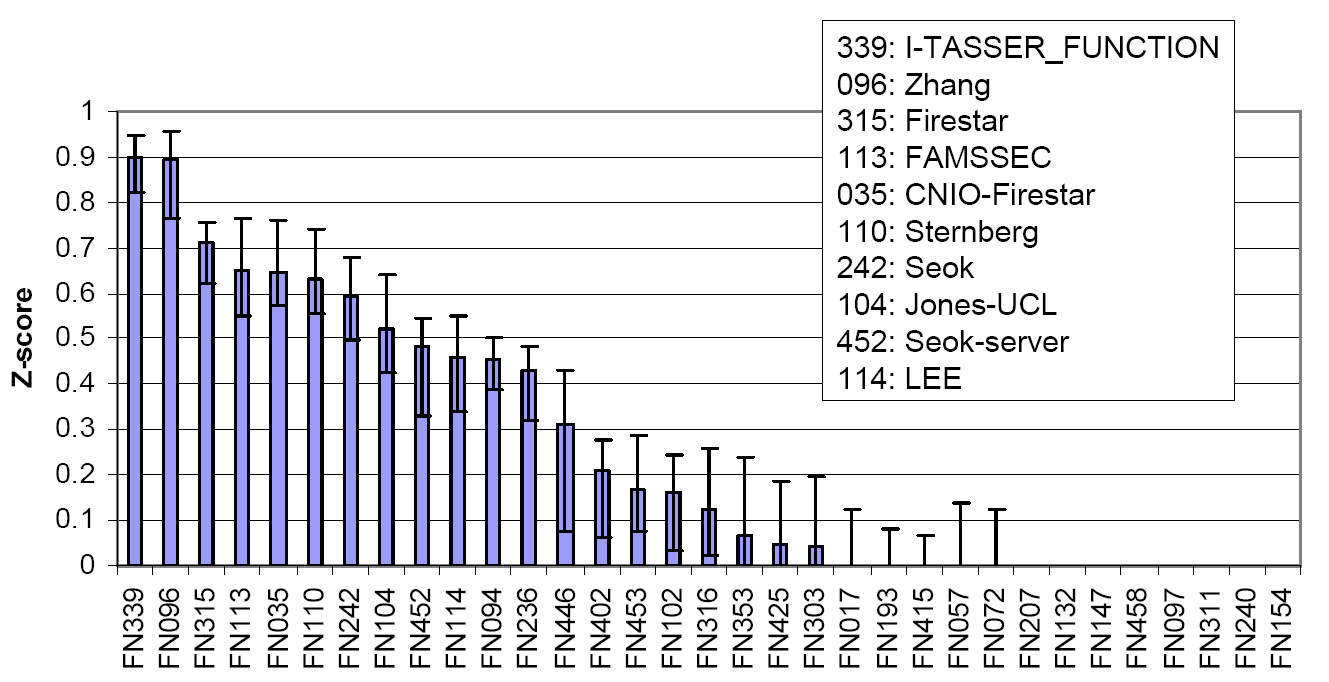
The COFACTOR was tested in the community-wide CASP9 experiment as "I-TASSER_FUNCTION" in the Server section and as "ZHANG" in the Human section, which were ranked at the first two positions in both Z-score and the Matthews correlation coefficient (MCC) compared with the experimental data (Figure 6).
|
SNP Mutation and Genetic Disease
|

We have recently studied the impact of SNP mutations on the protein folding stability, and found that the SNP-induced free-energy changes (i.e., ddG, Figure 8A), calculated from protein structure prediction, are closely correlated with the experimental measurement, demonstrating the feasibility of using low-resolution structure prediction information to examine the effect of gene mutations (Figure 8B). In another study, we investigated the impact of SNP mutations on the stability of protein-protein interactions (PPI). It was found that the interface structural profiles, collected from homologous PPI interfaces, can be used to accurately calibrate the changes of protein-protein binding affinity by SNP mutations (Figure 8C).
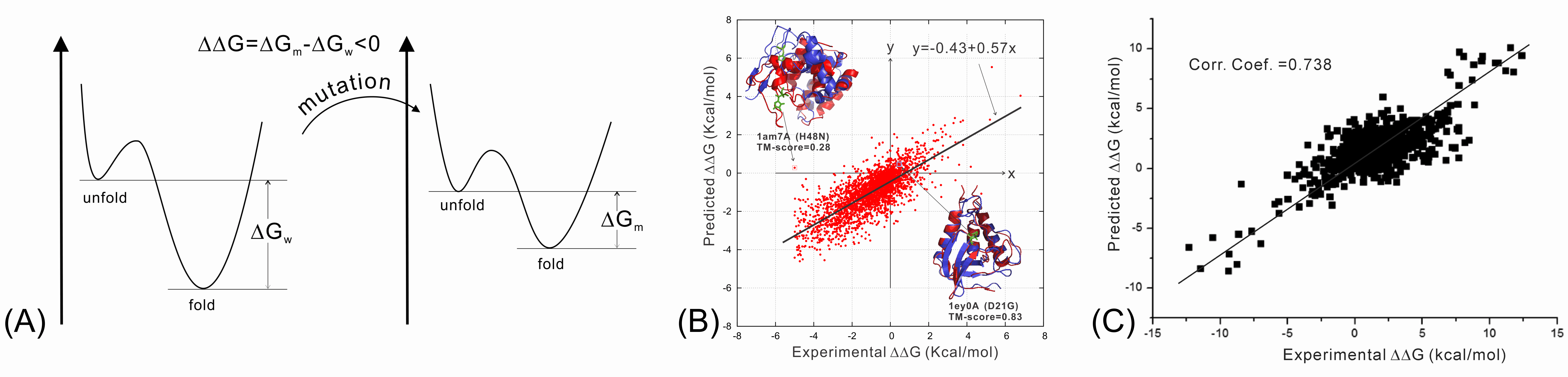
We are now working on the use of protein structure modeling techniques to predict what mutations would and what would not cause human diseases (in particular cancer). One goal is to deduce the quantitative relation of SNP mutations and specific human diseases, which should significantly enhance the impact of protein structure prediction on human disease diagnosis and treatment studies.
|
Modeling of Protein-Protein Interactions
|

Figure 9. Rhodopseudomonas palustris protein-protein interaction network.
To predict 3D structure of protein-protein complexes from sequence, we developed a new dimeric threading algorithm, COTH, to recognize template structure of protein complexes from solved complex structural databases. COTH aligns multiple-chain sequences simultaneously through the PDB library using scoring functions including multiple sequence profiles and structural information, with the assistance of interface predictions from BSpred. The COTH algorithm demonstrated significant advantage compared to other homology-based template identification methods (Figure 10).

Figure 10. TM-score of templates identified by COTH versus that from other homology-based methods.
Following COTH, we are working on the development of Dimer-ITASSER by extending the I-TASSER algorithm for multiple-chain full-length complex structure prediction. Since the folding principles of protein domains and complexes are essentially the same, we are hopeful to exploit I-TASSER iterative threading assembly methodology to significantly refine the template structures as identified from COTH, with the focus on the modeling of binding-induced side-chain and backbone conformational changes. One of the long-term goals is to utilize the developed Dimer-ITASSER to reconstruct the structure-based protein-protein network across genomes. A systematic, atom-level description of protein-protein complexes will be essential for the understanding of cellular processes and for the development of novel reagents to regulate the protein-protein interaction networks.
|
G Protein-Coupled Receptor and
Ligand-Receptor Interactions
|
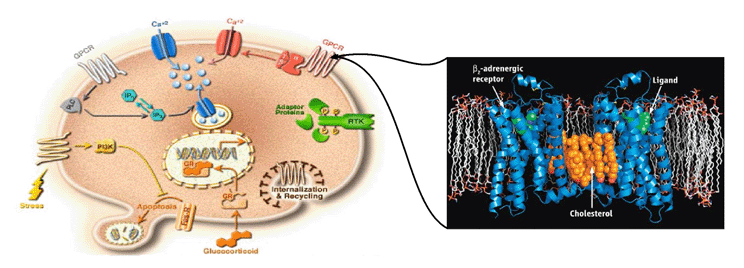
We are working on the development of the new GPCR modeling tool, GPCR-ITASSER, which extends I-TASSER by incooporating the protein-membrane interactions and the mutagenesis restraints into the knowledge-based force field. The ligand-GPCR interactions are then modeled by BSP-SLIM, a blind molecular docking tool designed for low-resolution protein-ligand docking. The method was tested (as "UMich-Zhang") in the recent community-wide GPCR Dock experiment in 2010. Figure 12 shows the result of our lab on all three ligand-GPCR complexes, where the first receptor models are 2.4 and 1.6 Angstroms to the crystal structure in the transmembrane region for the CXCR4 chemokine and dopamine D3 receptors, respectively. The three ligands, antagonists IT1t, CVX15, and eticlopride, are all in the same pocket as that in the crystal structure (Figure 12).
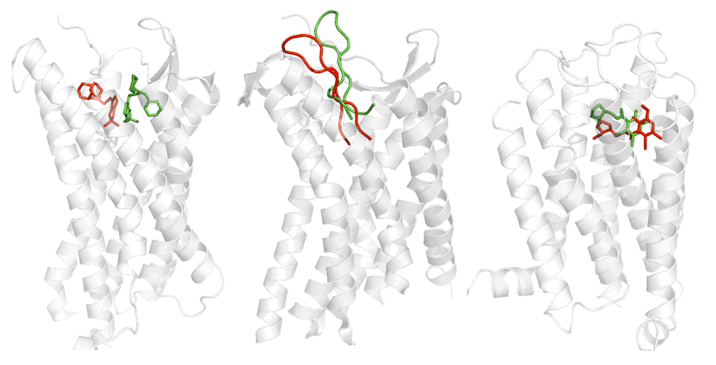
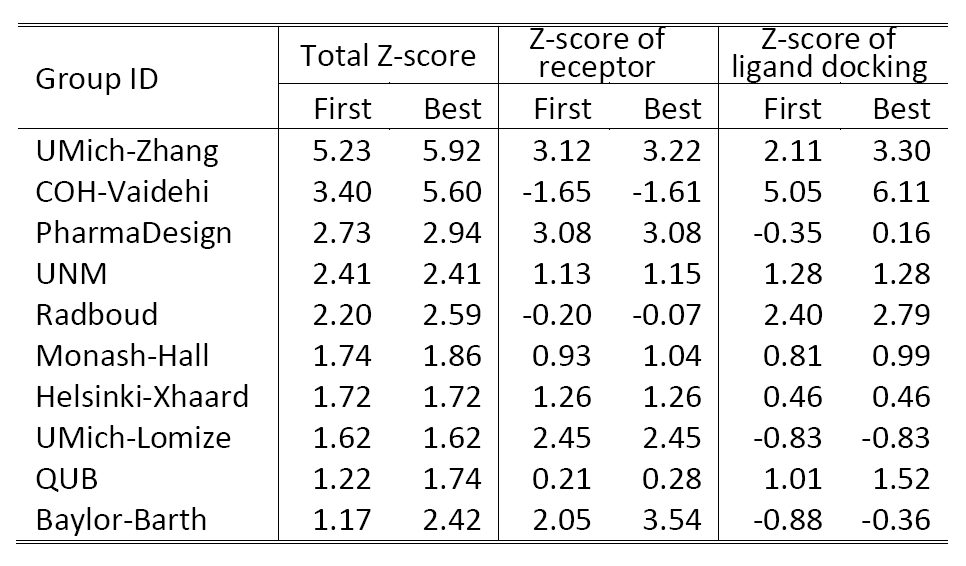
Table 2 shows a summary of the top 10 groups (out of 35) in GPCRDock 2010, together with the cumulative Z-score on all three targets for both receptor and ligand models. The most significant success of our models is on the distant homology target CXCR4/CVX15, as Kufareva et al. (the assessors) commented, "Modeling the CXCR4/CVX15 peptide complex represented the biggest challenge of GPCR Dock 2010. The top model of this complex (by UMich-Zhang) has the Z-score of 2.45 thus far exceeding other models in accuracy."
We are now working on the application of the GPCR-ITASSER and BSP-SLIM pipeline to the modeling of all GPCRs in the human genome, to generate high-resolution structures of the receptors as well as the ligand-receptor associations with the aid of experimental restraints collected in GPCRRD. One goal is to systematically annotate the physiological roles of all GPCRs in associated pathways and to identify new therapies to regulate these interactions.
|
Ligand Screening and Structure-Based Drug Design
|

Figure 13. A successful example of structure-based drug design by Bugg et al. in 1990s
in designing a molecule
that inhibits enzyme purine nucleoside phosphorylase (PNP). PNP normally takes up individual nucleosides (a)
and cleaves the purine from the sugar, giving rise to a free purine base and a phosphorylated sugar (b).
A tightly fitting compound blocks the binding pocket and therefore inhibits the acitivity of the PNP enzyme (c).
We recently developed a composite approach for druglike compound identification, which combines structure-based virtual screening with quantitative structure-activity relationship (QSAR). When using the approach to the epidermal growth factor receptor (EGFR), an important target protein associated with brain, lung, bladder and colon tumors, we found that two compounds (2 and 21) have significant EGFR-inhibitory activities (Figure 14). The experimental assay to test the ability of the compounds in inhibiting the receptor proteins is in progress.

Figure 14.
Binding structure of two compounds screened from the ZINC library which have inhibitory
activity on the epidermal growth factor receptor (EGFR), an important tumor target protein.
yangzhanglab![]() umich.edu
| (734) 647-1549 | 100 Washtenaw Avenue, Ann Arbor, MI 48109-2218
umich.edu
| (734) 647-1549 | 100 Washtenaw Avenue, Ann Arbor, MI 48109-2218