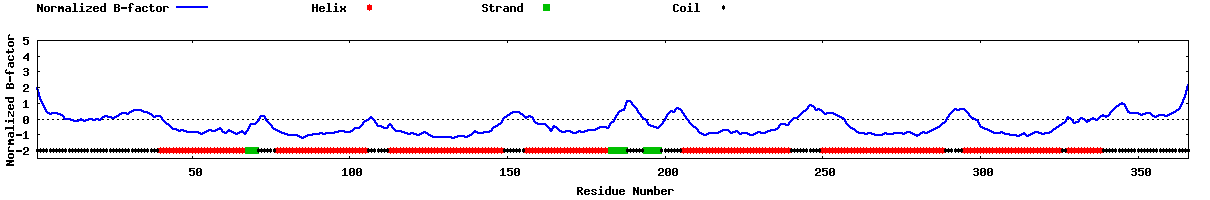
| 20 40 60 80 100 120 140 160 180 200 220 240 260 280 300 320 340 360 | |
| | | | | | | | | | | | | | | | | | | | |
| MWNATPSEEPGFNLTLADLDWDASPGNDSLGDELLQLFPAPLLAGVTATCVALFVVGIAGNLLTMLVVSRFRELRTTTNLYLSSMAFSDLLIFLCMPLDLVRLWQYRPWNFGDLLCKLFQFVSESCTYATVLTITALSVERYFAICFPLRAKVVVTKGRVKLVIFVIWAVAFCSAGPIFVLVGVEHENGTDPWDTNECRPTEFAVRSGLLTVMVWVSSIFFFLPVFCLTVLYSLIGRKLWRRRRGDAVVGASLRDQNHKQTVKMLAVVVFAFILCWLPFHVGRYLFSKSFEPGSLEIAQISQYCNLVSFVLFYLSAAINPILYNIMSKKYRVAVFRLLGFEPFSQRKLSTLKDESSRAWTESSINT | |
| CCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCHHHHHHHHHHHHHHHHHHHHHHHHHHHHSSSCCCCCCHHHHHHHHHHHHHHHHHHHHHHHHHHHHHCCCCCCCHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHCCCCCCCHHHHHHHHHHHHHHHHHHHHHHHHHHHSSSSSCCCCCCSSSSSCCCCCCCHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHCCCCCCCCCCHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHCCCCCCHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHCCHHHHHHHHHHHCCCCCCCCCCCCCCCCCCCCCCCCCCCC | |
| 989999999988788677788889888876632334575179999999999999999987896523224079998599999999999999999972799999982799889529888999999999999999999999983889972323030410777874014999999999999999825888268753023343588862277889997899999999999999999999999982657999751146888876499999988769999997999999999997457775148899999999999999999999999999829999999999838778777857887888888868999999 |
| 20 40 60 80 100 120 140 160 180 200 220 240 260 280 300 320 340 360 | |
| | | | | | | | | | | | | | | | | | | | |
| MWNATPSEEPGFNLTLADLDWDASPGNDSLGDELLQLFPAPLLAGVTATCVALFVVGIAGNLLTMLVVSRFRELRTTTNLYLSSMAFSDLLIFLCMPLDLVRLWQYRPWNFGDLLCKLFQFVSESCTYATVLTITALSVERYFAICFPLRAKVVVTKGRVKLVIFVIWAVAFCSAGPIFVLVGVEHENGTDPWDTNECRPTEFAVRSGLLTVMVWVSSIFFFLPVFCLTVLYSLIGRKLWRRRRGDAVVGASLRDQNHKQTVKMLAVVVFAFILCWLPFHVGRYLFSKSFEPGSLEIAQISQYCNLVSFVLFYLSAAINPILYNIMSKKYRVAVFRLLGFEPFSQRKLSTLKDESSRAWTESSINT | |
| 734314364132211132132322333323334323222111100001010100110231120000000003402100000010003001000000121001002333010010011001001100000000000000113100000002023313331000000001000000010000002023363343220000002223343121000011133312310100020001001101333446445534443432100000000000000000222001000000013344231220000011201000020001100000001430040023003023344443443454334344444468 |
| Rank | PDB Hit | Iden1 | Iden2 | Cov. | Norm. Z-score | Download Align. | 20 40 60 80 100 120 140 160 180 200 220 240 260 280 300 320 340 360 | | | | | | | | | | | | | | | | | | | |||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Sec.Str Seq | CCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCHHHHHHHHHHHHHHHHHHHHHHHHHHHHSSSCCCCCCHHHHHHHHHHHHHHHHHHHHHHHHHHHHHCCCCCCCHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHCCCCCCCHHHHHHHHHHHHHHHHHHHHHHHHHHHSSSSSCCCCCCSSSSSCCCCCCCHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHCCCCCCCCCCHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHCCCCCCHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHCCHHHHHHHHHHHCCCCCCCCCCCCCCCCCCCCCCCCCCCC MWNATPSEEPGFNLTLADLDWDASPGNDSLGDELLQLFPAPLLAGVTATCVALFVVGIAGNLLTMLVVSRFRELRTTTNLYLSSMAFSDLLIFLCMPLDLVRLWQYRPWNFGDLLCKLFQFVSESCTYATVLTITALSVERYFAICFPLRAKVVVTKGRVKLVIFVIWAVAFCSAGPIFVLVGVEHENGTDPWDTNECRPTEFAVRSGLLTVMVWVSSIFFFLPVFCLTVLYSLIGRKLWRRRRGDAVVGASLRDQNHKQTVKMLAVVVFAFILCWLPFHVGRYLFSKSFEPGSLEIAQISQYCNLVSFVLFYLSAAINPILYNIMSKKYRVAVFRLLGFEPFSQRKLSTLKDESSRAWTESSINT | |||||||||||||||||||||||||
| 1 | 4n6hA | 0.23 | 0.25 | 0.91 | 3.04 | Download | LANEGKVKEAQAAAEQLKTTRNAYIQKYLGSPGARSASSLALAIAITALYSAVCAVGLLGNVLVMFGIVRYTKMKTATNIYIFNLALADALATSTLPFQSAKYLME-TWPFGELLCKAVLSIDYYNMFTSIFTLTMMSVDRYIAVCHPVKALDFRTPAKAKLINICIWVLASGVGVPIMVMAVTRPRDGAVVCMLQF----PSPSWYWDTVTKICVFLFAFVVPILIITVCYGLMLLRLRSVRLLSGS---KEKDRSLRRITRMVLVVVGAFVVCWAPIHIFVIVWTLVDIDRR---DPLVVAALHLCIALGYANSSLNPVLYAFLDENFKRCFRQLCRKPCG----------------------- | |||||||||||||||||||
| 2 | 5vblA | 0.21 | 0.22 | 0.82 | 3.88 | Download | --------------------------------------------LIPAIYMLVFLLGTTGNGLVLWTVFRSREKRRSADIFIASLAVADLTFVVTLPLWATYTYRDYDWPFGTFFCKLSSYLIFVNMYASAFCLTGLSFDRYLAIVRPVANARLRLRVSGAVATAVLWVLAALLAMPVMVLRTTGDLENTNKVQCYMDYSMVASEWAWEVGLGVSSTTVGFVVPFTIMLTCYFFIAQTIAMKPLCGVGKDQFEEVEERRRLLSIIVVLVVTFALCKMPYHLVKTLYMLGLLHWPCDFDLFLMNIFPYCTCISYVNSCLNPFLYAFFDPRFRQACTSMLLMGQSR---------------------- | |||||||||||||||||||
| 3 | 4n6hA | 0.24 | 0.24 | 0.82 | 3.71 | Download | ------------------------------SPGARSASSLALAIAITALYSAVCAVGLLGNVLVMFGIVRYTKMKTATNIYIFNLALADALATSTL-PFQSAKYLMETWPFGELLCKAVLSIDYYNMFTSIFTLTMMSVDRYIAVCHPVKALDFRTPAKAKLINICIWVLASGVGVPIMVMAVTRPRDG-----AVVCMLQFPPSWYWDTVTKICVFLFAFVVPILIITVCYGLMLLRLRSVRLLSGS---KEKDRSLRRITRMVLVVVGAFVVCWAPIHIFVIVWTLVDIDRRDP---LVVAALHLCIALGYANSSLNPVLYAFLDENFKRCFRQLCRKPCG----------------------- | |||||||||||||||||||
| 4 | 4djh | 0.26 | 0.26 | 0.79 | 1.53 | Download | --------------------------------------SPAIPVIITAVYSVVFVVGLVGNSLVMFVIIRYTKMKTATNIYIFNLALADALVTTTMPFQSTVYL-MNSWPFGDVLCKIVLSIDYYNMFTSIFTLTMMSVDRYIAVCHPVKALDFRTPLKAKIINICIWLLSSSVGISAIVLGGTKVRE---DVDVIECSLQFPDDDYWWDLFKICVFIFAFVIPVLIIIVCYTLMILRLKSVRLLSGNIPYREKDRNLRRITRLVLVVVAVFVVCWTPIHIFILVEALG--SAA-------LSSYYFCIALGYTNSSLNPILYAFLDENFKRCFRDFCFP-------------------------- | |||||||||||||||||||
| 5 | 4djh | 0.25 | 0.26 | 0.80 | 1.16 | Download | --------------------------------------SPAIPVIITAVYSVVFVVGLVGNSLVMFVIIRYTKMKTATNIYIFNLALADALVTTTMPFQSTVYLM-NSWPFGDVLCKIVLSIDYYNMFTSIFTLTMMSVDRYIAVCHPVKALDFRTPLKAKIINICIWLLSSSVGISAIVLGGTKVREDVDVIECSLQFPDD-DYSWWDLFMKICVFIFAFVIPVLIIIVCYTLMILRLKSVRLLSGNTTYREKDRNLRRITRLVLVVVAVFVVCWTPIHIFILVEALGSA---------ALSSYYFCIALGYTNSSLNPILYAFLDENFKRCFRDFCFP-------------------------- | |||||||||||||||||||
| 6 | 4n6hA | 0.25 | 0.24 | 0.82 | 3.43 | Download | --------------------------------GARSASSLALAIAITALYSAVCAVGLLGNVLVMFGIVRYTKMKTATNIYIFNLALADALATSTLPFQSAKYLM-ETWPFGELLCKAVLSIDYYNMFTSIFTLTMMSVDRYIAVCHPVKALDFRTPAKAKLINICIWVLASGVGVPIMVMAVTRPRDGAVVCMLQF----PSPSWYWDTVTKICVFLFAFVVPILIITVCYGLMLLRLRSVRLL---SGSKEKDRSLRRITRMVLVVVGAFVVCWAPIHIFVIVWTLVDIDRR---DPLVVAALHLCIALGYANSSLNPVLYAFLDENFKRCFRQLCRKPCG----------------------- | |||||||||||||||||||
| 7 | 4djh | 0.26 | 0.26 | 0.78 | 1.69 | Download | -----------------------------------------IPVIITAVYSVVFVVGLVGNSLVMFVIIRYTKMKTATNIYIFNLALADALVTTTMPFQSTVYL-MNSWPFGDVLCKIVLSIDYYNMFTSIFTLTMMSVDRYIAVCHPVKALDFRTPLKAKIINICIWLLSSSVGISAIVLGGTKVREDVDVIECSLQFPDDDY-SWWDLFMKICVFIFAFVIPVLIIIVCYTLMILRLKSVRLLSGNDAYREKDRNLRRITRLVLVVVAVFVVCWTPIHIFILVEALGSA---------ALSSYYFCIALGYTNSSLNPILYAFLDENFKRCFRDFCF--------------------------- | |||||||||||||||||||
| 8 | 4n6hA | 0.25 | 0.24 | 0.83 | 4.75 | Download | ----------------------GSPG-------ARSASSLALAIAITALYSAVCAVGLLGNVLVMFGIVRYTKMKTATNIYIFNLALADALATSTLPFQSAKYLME-TWPFGELLCKAVLSIDYYNMFTSIFTLTMMSVDRYIAVCHPVKALDFRTPAKAKLINICIWVLASGVGVPIMVMAVTRPRDG----AVVCMLQFPSPSWYWDTVTKICVFLFAFVVPILIITVCYGLMLLRLRSVRLLSGS---KEKDRSLRRITRMVLVVVGAFVVCWAPIHIFVIVWTLVDIDRRD---PLVVAALHLCIALGYANSSLNPVLYAFLDENFKRCFRQLCRKPCG----------------------- | |||||||||||||||||||
| 9 | 4n6hA | 0.24 | 0.24 | 0.83 | 3.35 | Download | ------------------------------SPGARSASSLALAIAITALYSAVCAVGLLGNVLVMFGIVRYTKMKTATNIYIFNLALADALATSTLPFQSAKYLM-ETWPFGELLCKAVLSIDYYNMFTSIFTLTMMSVDRYIAVCHPVKALDFRTPAKAKLINICIWVLASGVGVPIMVMAVTRPRDGAVVCMLQF----PSPSWYWDTVTKICVFLFAFVVPILIITVCYGLMLLRLRSVRLLSGS---KEKDRSLRRITRMVLVVVGAFVVCWAPIHIFVIVWTLVDIDRR---DPLVVAALHLCIALGYANSSLNPVLYAFLDENFKRCFRQLCRKPCG----------------------- | |||||||||||||||||||
| 10 | 5c1mA | 0.24 | 0.23 | 0.81 | 4.31 | Download | ---------------------------GSHSLPQTGSPSMVTAITIMALYSIVCVVGLFGNFLVMYVIVRYTKMKTATNIYIFNLALADALATSTLPFQSVNYL-MGTWPFGNILCKIVISIDYYNMFTSIFTLCTMSVDRYIAVCHPVKALDFRTPRNAKIVNVCNWILSSAIGLPVMFMATTKYRQGSIDCTLTF----SHPTWYWENLLKICVFIFAFIMPVLIITVCYGLMILRLKSVRMLSGS---KEKDRNLRRITRMVLVVVAVFIVCWTPIHIYVIIKALITIPET----TFQTVSWHFCIALGYTNSCLNPVLYAFLDENFKRCF-------------------------------- | |||||||||||||||||||
| ||||||||||||||||||||||||||
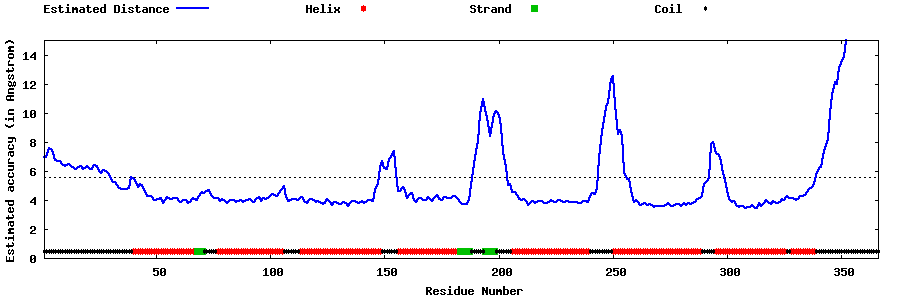
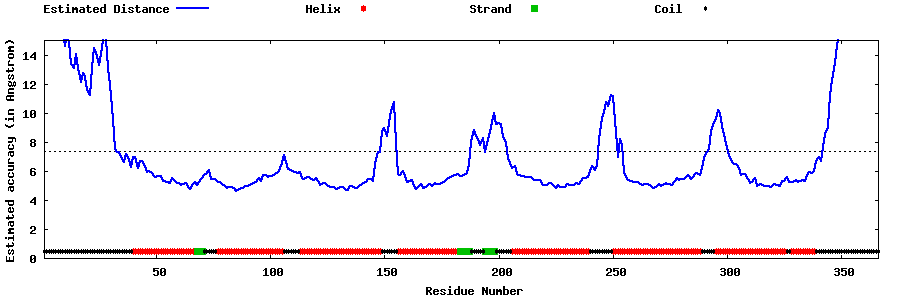
| Generated 3D models | Estimated local accuracy of models | ||
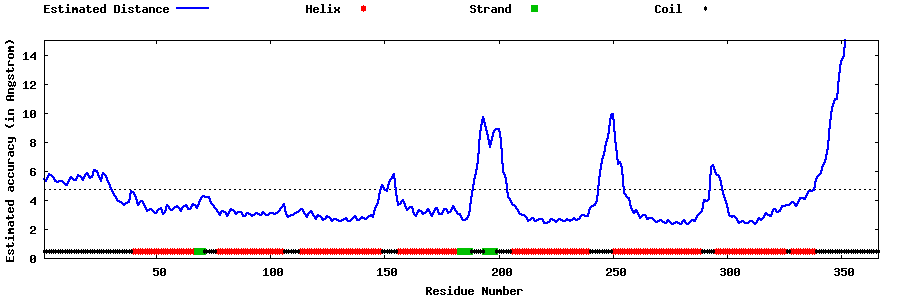 | |||
|
|
|||
 | |||
|
| |||
 | |||
|
| |||
 | |||
|
| |||
 | |||
|
| |||