

EvoDesign is an evolution-based approach to de novo protein fold and protein-protein interaction design.
It takes the full-atomic model of a scaffold in PDB format and outputs a list of designed sequences along
with the percent sequence identity of each sequence to the starting scaffold. EvoDesign also provides
normalized relative errors for predicted secondary structure, solvent accessibility, and backbone torsional
angles with respect to the input. This helps the user understand the quality of the designed sequences.
| EvoDesign Overview |
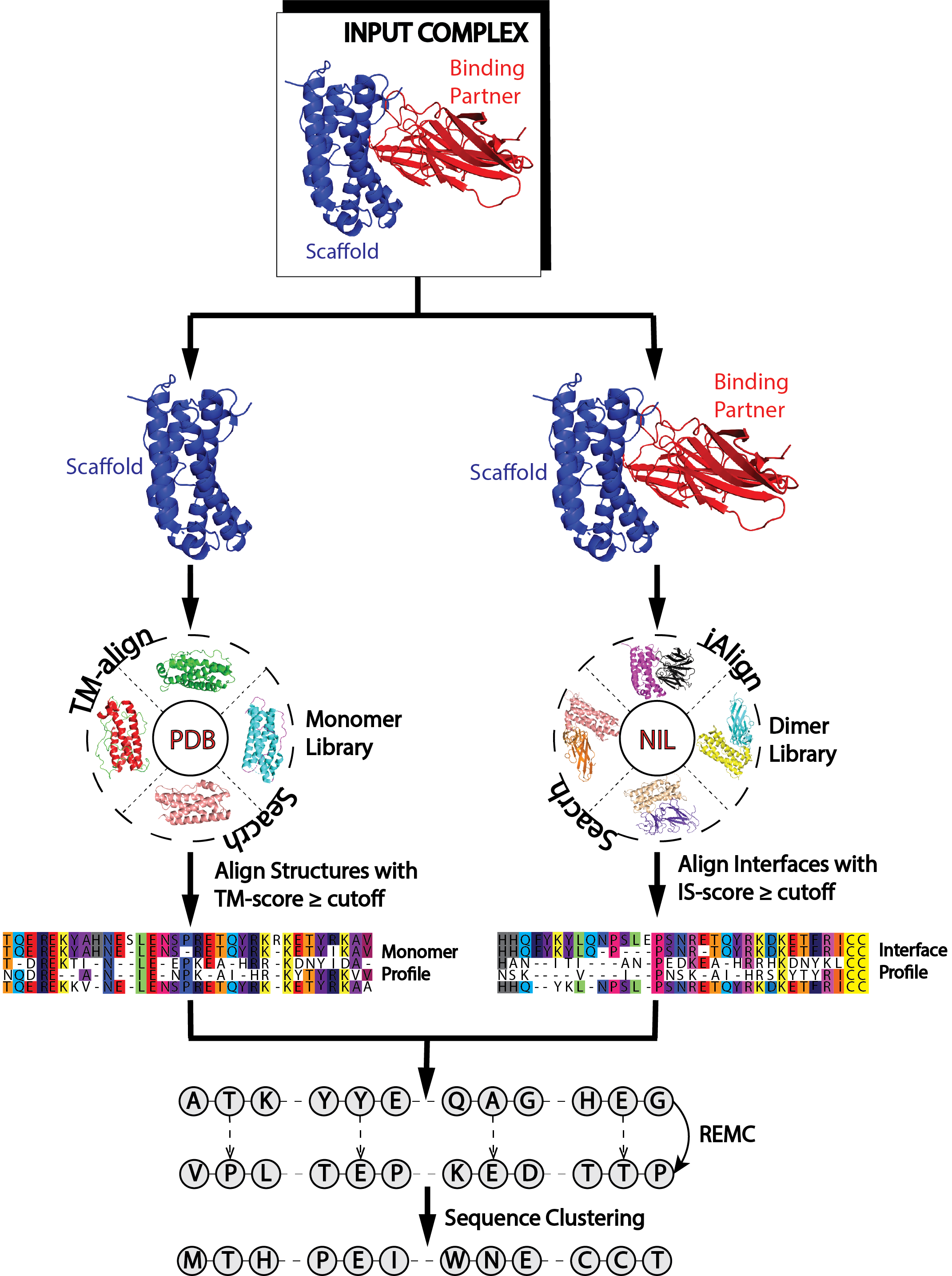
Figure 1. EvoDesign pipeline. |
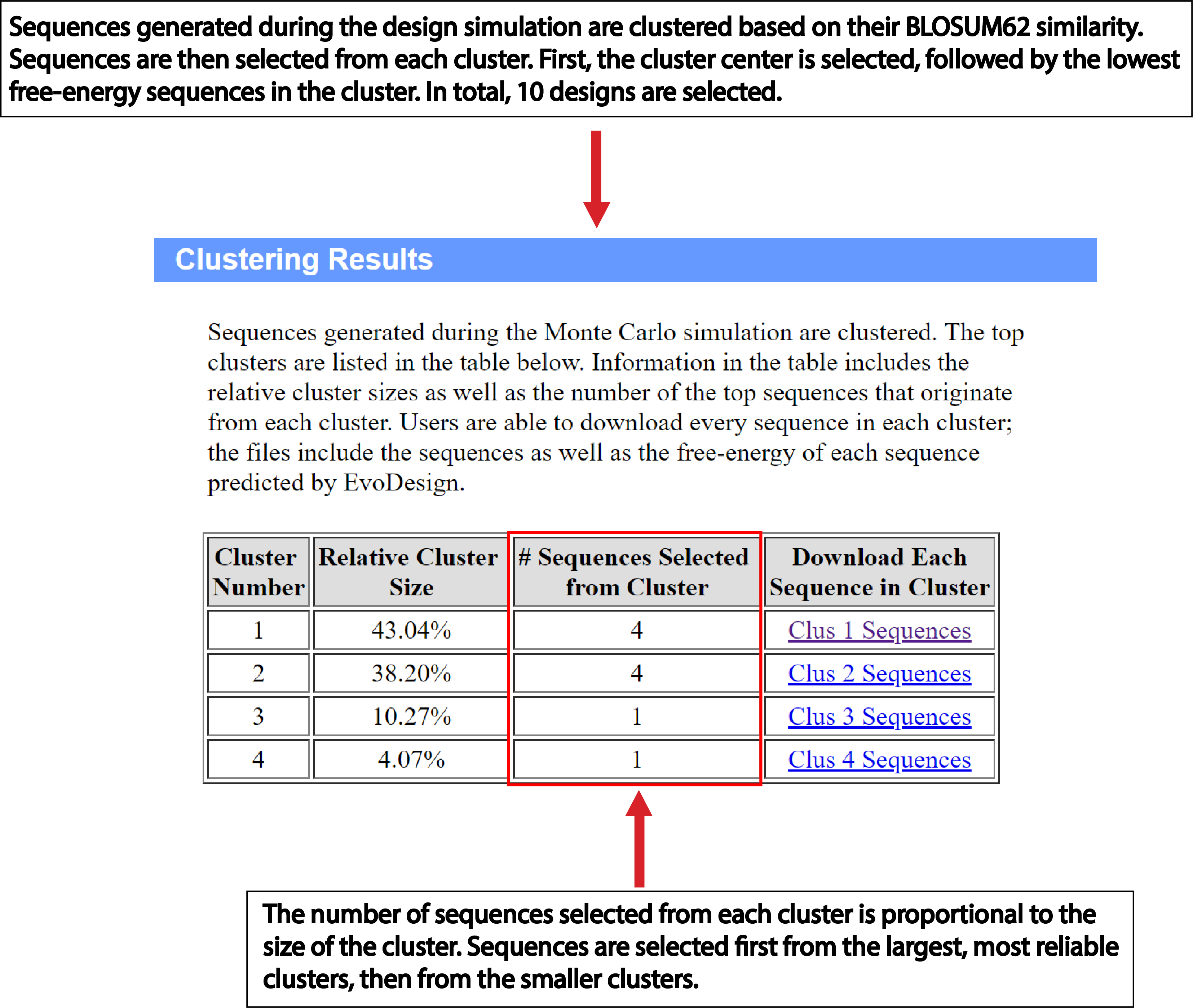
EvoDesign has two design options: monomer and protein-protein interface design. Both design strategies consist of three stages – pre-processing, simulation, and analysis of the data generated during the simulation phase to produce the final designed sequences (Fig. 1). Pre-processing: For monomer design, the program starts with a given scaffold and searches the Protein Data Bank (PDB) for proteins with similar folds using our in-house TM-align program [1]. The primary structures of proteins with similar folds are then aligned to generate a profile. DSSP is used to assign secondary structure, solvent accessibility and backbone torsional angles for the scaffold. For protein interface design, the user must not only supply the scaffold of interest, but also its protein binding partner in PDB format. From here, a dimeric profile is constructed from multiple structure alignments of protein-protein interfaces with high similarity to the input complex. Simulation: The core of EvoDesign is a Monte Carlo (MC) based sequence space search. MC simulations start from a random seed sequence. Then replica-exchange Monte Carlo is used by EvoDesign to search for the lowest free-energy sequences. At each step of the simulation, the sequence is altered by mutating a number of randomly selected amino acids at randomly selected positions; then the energy of the sequence is computed. A sequence is accepted or rejected based upon the Metropolis criteria. Analysis: Sequences accepted during the Monte Carlo simulation are clustered using the SPICKER algorithm [2] following the same procedure as used by Bazzoli et al. [3]. The process works iteratively to identify the sequence with the maximum number of neighbors. EvoDesign outputs the top 10 sequences selected from the largest clusters. |
| Submitting an EvoDesign Job |
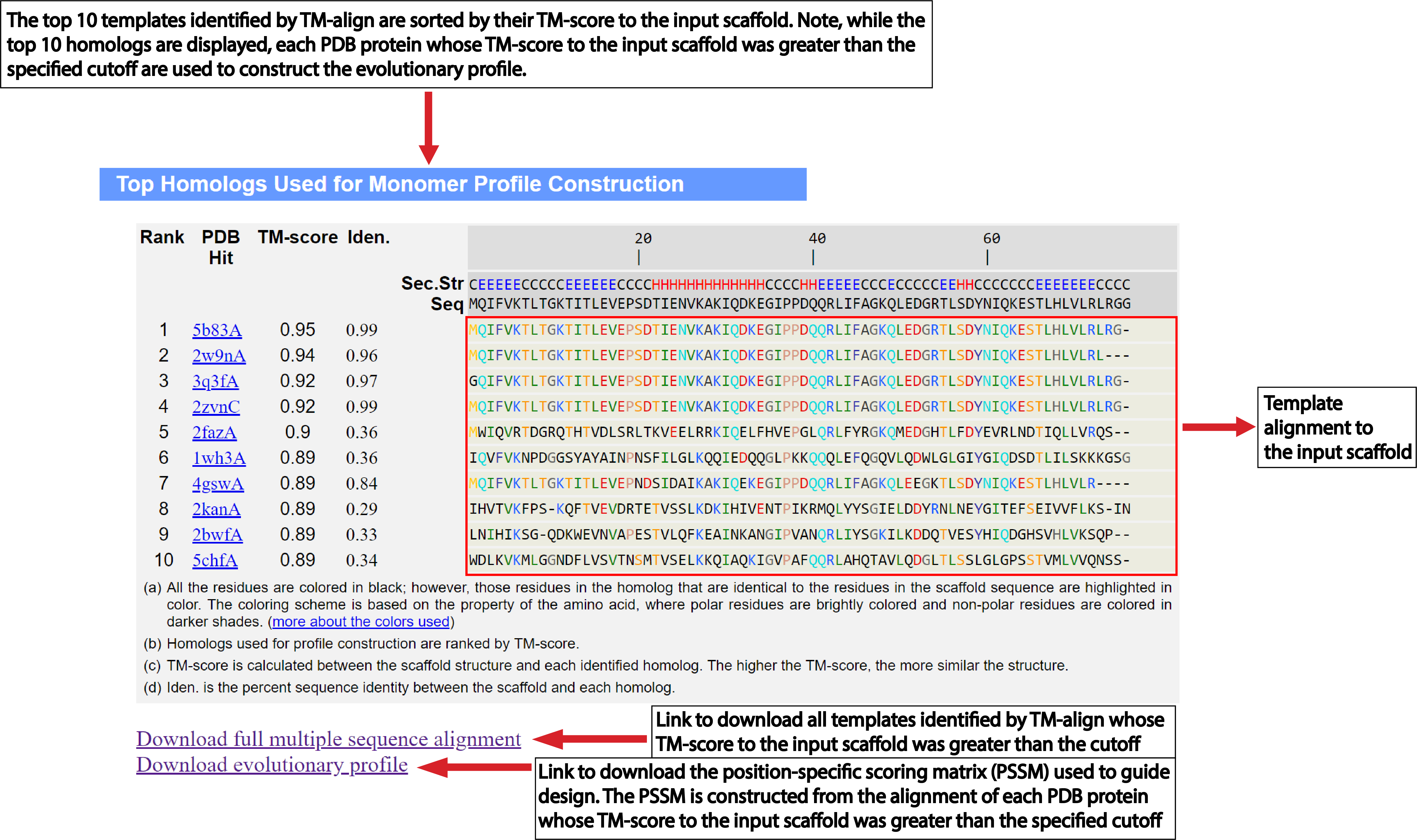
Structural profile cutoff:
By default, EvoDesign will look for highly similar folds in the PDB
(TM-score>0.7)
for structural profile
construction. Thus, the number of proteins in the profile may differ from one scaffold to another based
on their fold types. Again, it has been shown that to avoid sequence biasing during design
simulation, at least ten proteins are required [4].
Therefore, users may opt for a lower cutoff for profile construction if the fold of
the scaffold is novel. It should be noted that evolutionary information content decreases
with a decrease in TM-score cutoff [4]. Therefore, an optimized choice
is required for selection of this threshold.
Energy function:
For computational efficiency, the default energy setting for monomer design is to use the evolution-based
energy function alone. Our benchmarking validates the sufficiency of evolution-based energy functions to
design reasonably good sequences. Nonetheless, for experimental purposes, we suggest that users use both
the evolution and physics-based energy function. Furthermore, interface design requires the use of the
combined energy function. Note, the use of the evolution- and physics-based energy
function is 2-3 times slower than evolution-based only design. The excess time requirement is due to
computing intensive side chain refinement. A detailed discussion of the evolution-based energy function
can be found at Mitra et al. [4] and Pearce et al. [5].
It is important to note that the server no longer uses FoldX, but our own physical energy function that has been
optimized for our pipeline.
Restrictions on residues: Users can control the design by restricting one or more residues
type(s) (for instance users may wish to restrict
CYS in the design sequence) or by fixing some of the residues by inputting their residue ID.
Model designed sequences using
I-TASSER:
Users can model the designed sequences using I-TASSER by checking 'Yes' under the I-TASSER modeling option.
Since this step demands a great deal of computing resources, the default option is 'No'. We urge the
user to use this option properly. Alternatively, the user can use the
I-TASSER
web server to model the designed sequences.
Name of your protein: This is purely for inventory purposes. We suggest naming protein such that
users are able to discriminate
between different EvoDesign runs for different proteins.
Email: Email addresses are needed to send job completion notifications
with the link to the results page.
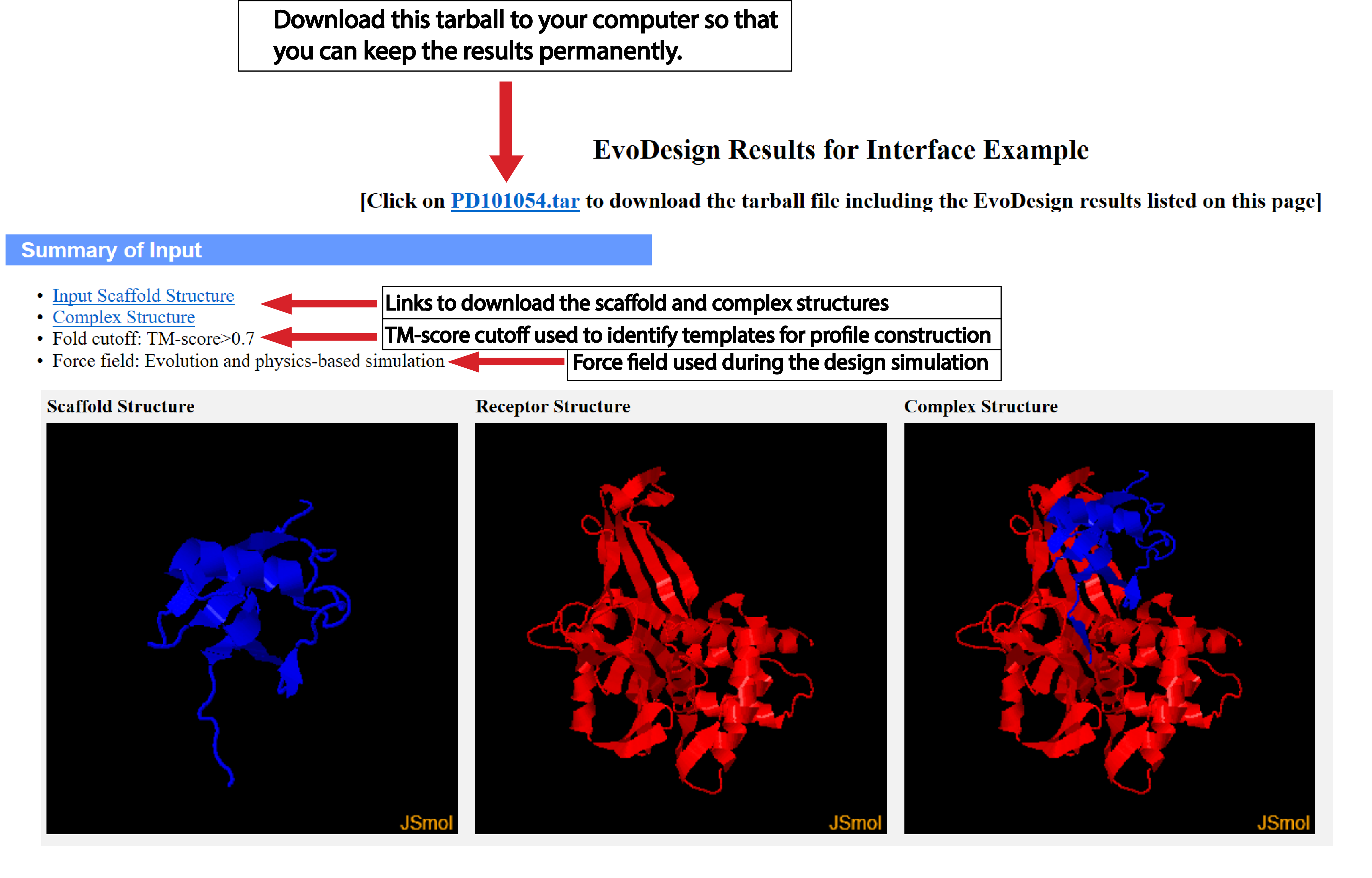
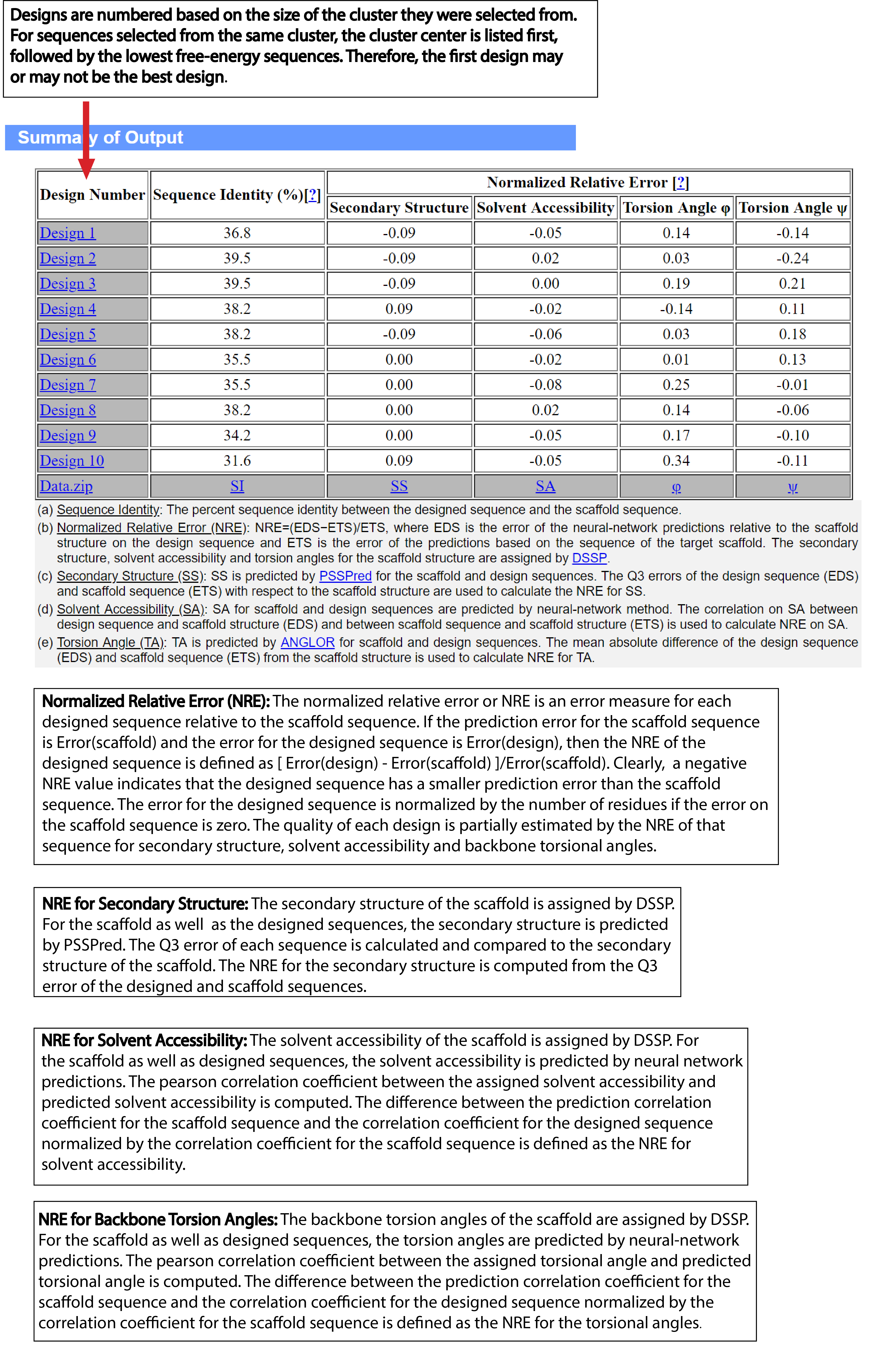
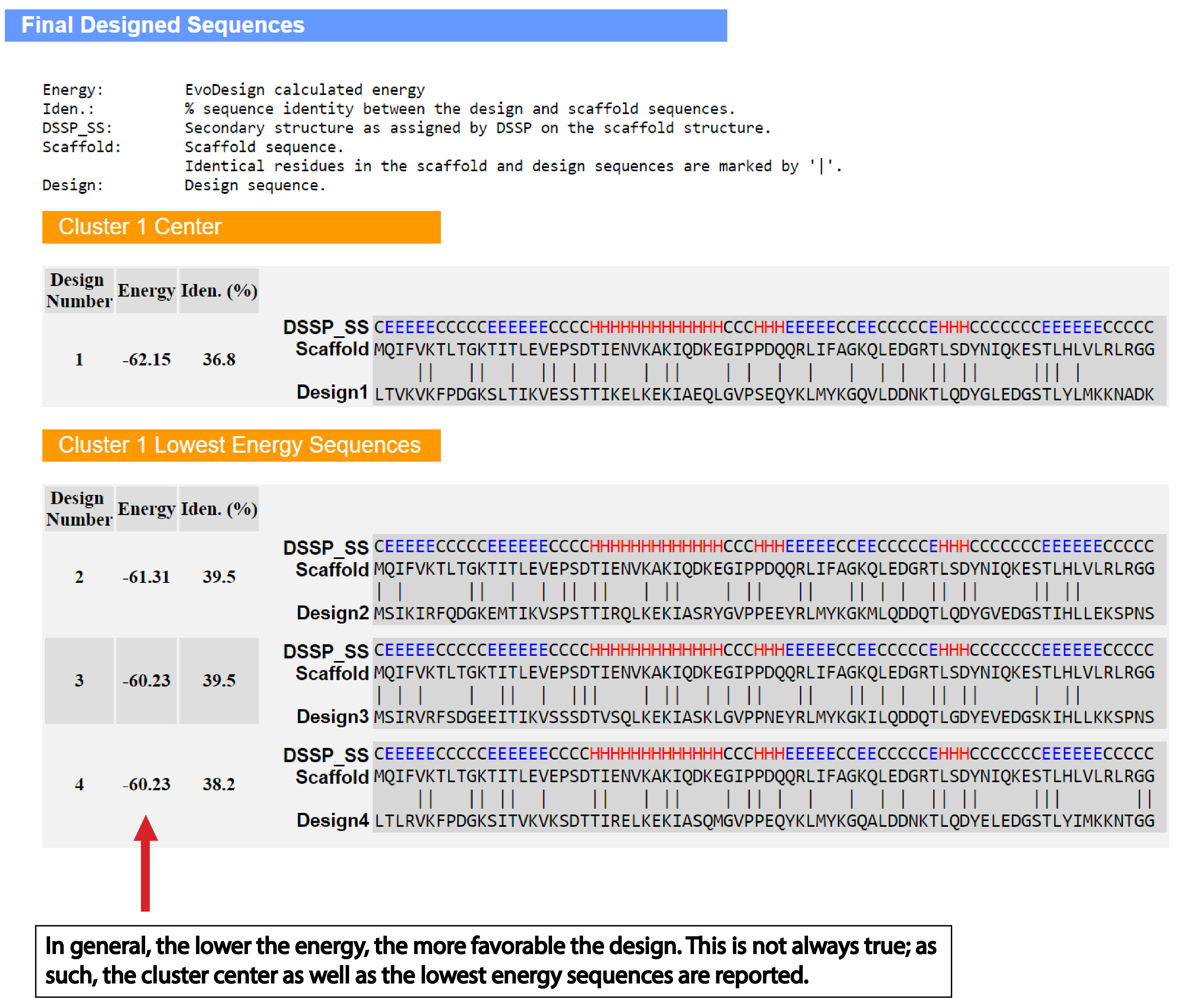
| Understanding the Output of EvoDesign |

yangzhanglab![]() umich.edu
| (734) 647-1549 | 100 Washtenaw Avenue, Ann Arbor, MI 48109-2218
umich.edu
| (734) 647-1549 | 100 Washtenaw Avenue, Ann Arbor, MI 48109-2218